When Erwin Schrödinger, famous for his “Schrödinger’s cat” thought experiment, was awarded the 1933 Nobel prize for physics, his notable quantum mechanics equation could only be solved for the simplest systems, not for real-world applications.
The advent of computational techniques to approximate solutions of the Schrödinger equation changed all that, making ab inito (“from first principles”) quantum mechanics calculations possible for large molecules and even proteins.
Ab initio quantum chemistry methods attempt to solve the electronic Schrödinger equation given the positions of the nuclei and the number of electrons in order to yield useful information such as electron densities, energies and other properties of the system. The ability to run these calculations has enabled theoretical chemists to study short-lived and reactive intermediates (high-energy molecules somewhere between reactants and products that don’t like to hang around very long) that are key to the understanding of chemical reactions but are almost impossible to study experimentally.
The importance of ab initio quantum chemistry methods resulted in another Nobel prize win, this time in Chemistry, awarded in 1998 to British theoretical chemists John Pople and Walter Kohn. Professor Pople was instrumental in the development of one of the most respected and widely used, general purpose computational chemistry software packages — Gaussian.
Gaussian has undergone continuous development for more than 40 years, culminating in the latest release of Gaussian 16 and the accompanying helper application, GaussView 6.
Ab initio quantum mechanics calculations are computationally intensive but Gaussian 16 allows jobs to be spread over many cores and make use of large amounts of memory, something that is an ideal fit with Awoonga and FlashLite, UQ’s high-throughput and data-intensive high-performance Linux compute clusters.
RCC has recently procured a Gaussian 16 and GaussView 6 site licence for UQ, and will install the software on all three UQ high-performance compute clusters, namely Tinaroo, FlashLite and Awoonga, in addition to the currently available previous version, Gaussian 09, which doesn’t have GaussView.
The site licence means any UQ researcher can use the HPC-installed software or install the software on their desktop or local cluster without having to buy their own licence. RCC/QCIF eResearch Analyst Dr Marlies Hankel will help systems administrators install the software on their local clusters.
The result is wider availability of the latest versions of Gaussian and GaussView, which boast additional features that users have been asking for, and greater convenience for the university’s researchers and students as they can analyse their data in the comfort of their office and be more productive.
One such researcher is organic chemist Dr Robert Reid, a Senior Research Fellow in the Institute for Molecular Bioscience (IMB). He and his students research the design and preparation of new medicines for diseases, including arthritis and cancer, and routinely use Gaussian to solve diverse chemical problems.
Their research using Gaussian could lead to new, valuable anti-inflammatory drugs and new medicines to fight bacterial and fungal infections. Their discoveries have recently been published in high-impact journals Nature and the Journal of the American Chemical Society.
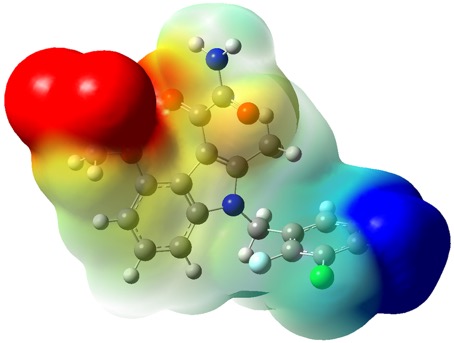
Gaussian calculations have also allowed Dr Reid and his team to determine how molecules will react by identifying their most electron-rich or deficient regions. They can also use the software to calculate the activation energy or barrier to a chemical process that determines whether it is likely to occur spontaneously or not. This can save researchers time by identifying the experiments that are most likely to work.
New chemical compounds either produced by nature, from plants or microbes, or synthesised in the laboratory could one day become new medicines. But to the eye they all look the same, usually white powders or clear liquids. So, chemists determine their composition using nuclear magnetic resonance spectroscopy (NMR) and then may use Gaussian to estimate the chemical shift parameters to confirm correct structural assignment. “We can visually compare the experimental NMR spectrum with the theoretically predicted spectrum using GaussView to see if they match and this has proven to be a very reliable method,” said Dr Reid.
The chemical problems that can be managed using Gaussian 16 have now become so intimately customisable that setting up the input file to run the job with the correct syntax had become “very tedious”, according to Dr Reid. Fortunately, this process is greatly simplified with the helper application GaussView, which arranges all the options in an easy-to-use graphical user interface.
The results from calculations that once were buried in large text files are also easily visualised using GaussView, which prepares graphical representations, tables (that can be exported to spreadsheets) and generates beautiful colour images (such as the above example in Figure 1, from Dr Reid, of the electrostatic surface potential of the molecule) in a high-quality publication-ready format.
“All of the world’s top universities have made Gaussian available to researchers, which obviously also includes The University of Queensland. We thank the RCC for continuing to keep us at the cutting edge by providing the very latest Gaussian release, which is such fantastic infrastructure to empower our research,” said Dr Reid.
If you would like to know more about Gaussian, please contact RCC’s Dr Marlies Hankel: m.hankel@uq.edu.au. For general enquiries about RCC’s HPCs, please contact the RCC Support Desk: rcc-support@uq.edu.au.

